


AI makes enzymes easier to engineer, unlocking new strategies for improving enzyme activity, stability or substrate specificity. Jake reviews a particularly challenging AI-guided enzyme engineering project from the Ginkgo foundry, highlighting the ways that AI is changing the shape of enzyme R&D projects.
Transcript
Today we're taking a closer look at a project that we did here at Ginkgo Bioworks using machine learning for enzyme engineering.
I love this one for a few reasons. First, it comes from the early days. We're really going into the vaults here - like 2 years ago. That's a long time in AI terms, long enough to get some perspective on how this worked and why.
Second, it shows how AI can break through when traditional approaches weren't getting there. Because we were working on an enzyme that our engineers call "recalcitrant." Basically this thing had a reputation for making synthetic biologists cry.
Let me break it down for you:
It was large, about 800 amino acids long.
Promiscuous, meaning it interacts with multiple different substrates.
Only partially characterized in terms of the mechanism of action.
All of these things tend to make an enzyme harder to engineer. But the challenge was worth it, because this enzyme also had an important role in central carbon metabolism. It belongs to one of these cycles inside a cell where carbon is shuffled around and converted like a currency exchange. That versatility makes it useful for many potential applications.
Our R&D partner wanted to improve the catalytic efficiency, essentially to make this thing run faster, without sacrificing other performance factors like the substrate affinity.
How did we do it? I want to break this project down into 4 generations. In each generation, we create new versions of the enzyme to design, build and test in the Ginkgo foundry. Selected candidates advance to become the basis for the next generation.
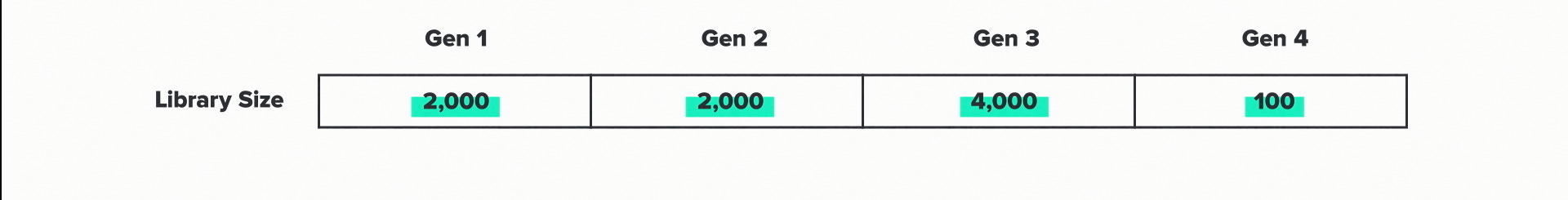
Each generation is associated with a library size, which represents the number of enzyme sequences that we tested. The more sequences that we try, the better chance we have of finding one with improved performance. The automation infrastructure at the foundry lets us test large numbers efficiently. But of course, data ain't free. So we're always looking for ways to design these libraries in ways that get more improvement from fewer tests.
Here are the library sizes for this project: 2000, 2000, 4000 and 100. This project was particularly challenging, so we tested more designs than we might normally. But the overall pattern here I think is going to be common in AI-driven biotech R&D. Large libraries at the beginning to train the models. Small libraries at the end, when the model-guided predictions start to get really good.
Each generation is also associated with one or more design strategies. The strategy varies depending on the unique features of the enzyme and the project. But in the big picture, they're all trying to solve the same basic problem: biological design space is too big to search completely.
Biology offers 20 amino acids for each position which, for an 800 amino acid protein, makes 20 to the power of 800 total possibilities which is one of those more-than-the atoms-in-the universe kind of numbers. A design strategy has to take us from effectively infinite possibilities to a testable number of options, ideally ones that are likely to improve performance.
Here's an example of a classical design strategy: active site mutagenesis. The active site is the structural pocket, usually deep inside the enzyme, where the chemical reaction takes place. The closer an amino acid is to this site, the more likely it is to influence the reaction. Obviously this isn't always the case, but it helps to focus the search.
A more sophisticated approach uses structural modeling software like Rosetta to describe the exact shape of the active site and where the substrate fits. We can select individual mutations to sample in Rosetta, then score them for how they affect the substrate binding. It's the classic lock-and-key situation, the enzyme and the substrate fit together, modeled with physics at atomic-scale resolution.
Active site mutagenesis and Rosetta docking are useful in the early stages, because they can be done computationally and don't require any new data. We also have AI strategies we can deploy without new data. Machine learning is famously a very data-intensive process. So you might think that we have to wait until we've built and tested some enzymes before we can start training the model. But there is a lot we can do with foundation models that use more general forms of biological data

.Evolutionary Scale Models are trained using essentially all known protein sequences. You can think of them as capturing the overall sequence properties that are common to naturally evolved proteins. Even though each position in a protein sequence can be one of 20 amino acids, they are not all equally likely. The probability of finding a given amino acid at a given position is influenced by its neighbors and by the sequence of the protein as a whole.

The global patterns in protein sequences are created by evolution. Evolutionary Scale Models can learn these patterns and then suggest changes to a protein that are evolution-like - changes that are more likely to be functionally important than random mutations would be.
That makes three strategies: Active site mutagenesis, Rosetta docking, Evolutionary Scale Models. Of course, these aren't mutually exclusive, they're overlapping. We can choose to select sequence changes that are evolution-like, in the enzyme active site, and improve the predicted docking.
In the first round, we sampled from all three - 2000 sequences total. We designed, built and tested all those designs. The result? A measly 50% improvement in catalytic efficiency.
We're we discouraged? Heck no. I'll be real with you: we usually want our gen 1 libraries to perform better than this. But remember - this enzyme had a reputation and we knew what we were getting into. So we pressed on.
In the second round, we were able to add the strategy of hit recombination. This means mixing and matching the best performing sequences from the first round. If mutation A gave us some improvement and mutation B gave us some improvement, what about a sequence with both A and B.
When we explore that gen 1 dataset, we're trying to balance optimality with diversity. We want to focus on the best performers, without completely closing the door on sequences that might open up new design spaces in the future.
All of this came together for the Gen 2 library. The result? 3.9X improvement. Respectable, but still not amazing. But now we had real data that we could bring into our most powerful machine learning tools: assay-labeled data.
Assay-labeled means we have a set of enzyme sequences and, for each one, a measurement of that enzyme's performance. Training an AI model with real data puts us much closer to the problem that we're trying to solve. Other strategies work from heuristics and proxy metrics. Active sites are probably important, evolution, docking. Assay labeled data gives us a ground truth and good model alignment, meaning we're optimizing directly for the measured performance that we want.
Our platform for handling assay-labeled data is called Owl, because it can see in the dark.
For Gen 3, we scored 4000 different sequences for their catalytic efficiency and their ability to express and fold. When I first heard about the Gen 3 library - I had kind of a Ginkgo moment. Like - we really just tested 4000 enzymes? We did. That's foundry-scale biological data.
The result in Gen 3 was a 4.5X improvement, only a little bit better than the previous round. But that's not the end of the story.
When I worked in the lab, back in the pre-AI era, we would often think about biological design in terms of diminishing returns. The first set of designs includes your most promising candidates, so it produces the most improvement. The second iteration is about making minor tweaks, and results in less improvement. By the third round, you're all out of good ideas and further improvements are marginal at best. Machine learning turns that on its head. Accumulating data means that your best designs are more likely to come in the later rounds.
We can see this effect by breaking out the gen 3 library by design strategy. The conventional strategies, active site mutagenesis and Rosetta docking, really were reaching the point of diminishing returns. They were giving us some improvements, but the bulk of the sequences were performing at or below expectations. The Owl-generated sequences, on the other hand, were not just producing the highest performers overall, they were consistently high. The AI platform wasn't just getting lucky once or twice, it was describing an entire design space where almost all the enzyme designs were good.

This is the mark of a well trained model. It only took 100 more designs to kick us all the way up to a 10x improvement in catalytic efficiency, our customer's target. AI was better than our conventional methods and better than any conventional approaches that we've seen used by others for this enzyme. AI got us there.

But really, data got us there. The AI tools for this project were built on two large datasets. The evolutionary scale models trained on millions of natural protein sequences. The assay-labeled models trained with thousands of experiments performed here in the Ginkgo foundry. AI made this enzyme easier to engineer because it made all that data more valuable.

“It had a reputation for making synthetic biologists cry” 😂